自宅でできる化学①~量子化学計算~
飛べない鳥
1.まえがき
皆さま、コロナ禍の中いかがお過ごしでしょうか。「はじめに」でも出てきた部長の飛べない鳥です。サークルの活動制限のためしばらく駒場の実験室に行くことができなくなり、学生実験もオンライン化してしまいました。やっぱり化学をしたいなあと思っていると、自分のノートパソコン上でも動かせるWinmostarなる量子化学計算ができるソフトウェアの存在を思い出し、「自宅でできる化学」と題して記事を書くことにしました。
2.Winmostarの導入
まずは計算環境を整えましょう。Winmostarの公式サイトにアクセスし、「学生版・無償版はこちら」をクリックします。メールアドレスを打ち込む欄があるので、在籍している教育機関のメールアドレスを記入します。中学生・高校生などで教育機関から配布されるメールアドレスを持っていない場合は教員を通じて申請するようにしましょう。あとはライセンス種類で「学生版」にチェックを入れて(無償版と学生版とで使用できる機能に差があります)、使用規約及びプライバシーポリシーに同意して登録。メールは登録して割とすぐに来ます。あとはメールに記載されたリンクからインストール手順が丁寧に説明されたページに飛べるので、そこに書いてあるとおりにインストールすればOKです。
あと、他にも「ソルバ(計算を実行するプログラムのこと)のインストールが必要だよ~」みたいなことが書かれていますが、デフォルトでMOPAC(後述)のソルバが入っています。この記事で使うのはMOPACのみなので他のソルバのインストールは皆さんの興味に応じて行ってみてください。
ちなみに、WinmostarはmacOS、Linuxは非対応です。仮想マシンを駆使するなどして使うことができるにはできるようなので、検討してみてはいかがでしょうか。
3.量子化学計算法の種類
量子化学計算の手法は以下の4種類に分かれています。
① 経験的分子軌道法
② 半経験的分子軌道法
③ 非経験的(ab initio)分子軌道法
④ 密度汎関数法(density functional theory: DFT)
この節では各々の手法を簡単に解説していきます。今すぐ計算がしたいよ! という方は次の節へどうぞ。色々と用語が出てきますが、ぶっちゃけ自分はどれもよく理解しているわけではないので詳しくは立ち入りません。へ~そうなんだ、くらいに思って頂ければ幸いです。
① 経験的分子軌道法
Hückel法、拡張Hückel法がこれに該当します。Hückel法については大学1,2年次に習うような最も単純な計算手法ですね。扱う分子が大きいものでなければ、手計算で事足ります。しかしながら、このHückel法はπ電子しか扱わないため、その適用範囲はπ共役系の分子に限られています。(Hückel法の詳細は〇〇の記事参照)拡張Hückel法では分子軌道を、分子を構成する原子の価電子が存在する原子軌道の一次結合で表す(LCAO近似)ことで、σ電子を含めた全ての結合性電子を考慮します。例えば、メタン分子の計算では水素原子の1s軌道と、炭素原子の2s, 2px, 2py, 2pz軌道が考慮されます。他にも重なり積分を無視しないなどHückel法とは異なる仮定をおいて計算されます。詳しくは文献[1]を参照してください。
② 半経験的分子軌道法
PPP法や前節で出てきたMOPACプログラムがこれに該当します。分子軌道の計算はHartree-Fock法(後述)で行われるのが基本です。半経験的分子軌道法では、このHartree-Fock法で行う際の計算時間を減らすため、計算の途中で現れる積分値を実験値と合うようなパラメータとして与えたり、計算方法を簡略化したりします。「半経験的」という言葉はここに由来しており、次に述べる非経験的分子軌道法ではこうした経験的パラメータや近似を行なわずに計算します。数千原子程度の分子でも、短い計算時間でその分子特性や反応性を評価できるのが魅力ですね。しかしながら、やはり計算精度はどうしても非経験的分子軌道法に劣ってしまいます。ただ、近年改良が進んでおり、計算対象によっては評価のある非経験的分子軌道法に匹敵する精度の計算結果を出しているようです。
PPP法では平面π電子系を扱い、共役長や色素分子の色と構造との関係の解明に使われます。MOPACはJ. J. P. Stewart博士により開発された世界的にも有名なソフトウェアで、拡張Hückel法のように分子中の各原子の価電子のみを扱います。今回では使用しませんが、教育機関に所属している方であればこちらのサイトから最新版のMOPAC2016が無償でダウンロードできます。Winmostarに入っているのはMOPAC6とMOPAC7で、いずれも2000年よりも前に作られた結構古いソフトなのですが、少し遊ぶ程度の目的なら問題ないでしょう。
③ 非経験的(ab initio)分子軌道法
非経験的分子軌道法では、半経験的分子軌道法で利用したパラメータや近似を使わずに計算します。ab initioとはラテン語で「初めから」を意味する言葉で、別名「ab initio分子軌道法」とも呼ばれます。計算の精度が高い分、その非経験性の代償として、計算時間が長いです。どれくらい長いかというと、計算時間が基底関数の数、すなわちLCAO近似で用いる原子軌道の数の4乗で増大するほど長いです。GAMESSプログラムやGaussianプログラムでこうした計算を行うことができます。Gaussianは有料ですが、GAMESSはアメリカのものが無償で利用できます。詳しい導入方法はWinmostarのサイトに書かれていますので、そちらを参照して下さい。
④ 密度汎関数法(density functional theory: DFT)
密度汎関数法は分子などの基底状態を、それを構成する電子の密度分布を試行関数にして計算する方法で、試行関数に波動関数を用いる分子軌道法とは別物となっています。非経験性の高いものから半経験的なものまで多様な汎関数(関数の関数)が計算に用いられているようです。GAMESSプログラムやGaussianプログラムでこうした計算を行うことができます。密度汎関数法は最近だとかなり一般的に用いられる計算方法のようで、以前研究室で実習を受けてGaussianを触らせてもらう機会があったときはこの密度汎関数法を使っていろいろな分子を計算していました。いくつか欠点はあるようですが、今後も改良されていくことでしょう。
4.Winmostarを使う利点
量子化学計算のプログラムを動かすにはCUIを使う、つまりコマンドプロンプトでいちいちコマンドを打ちながらソフトウェアに指示を出さねばならず、何も知らない人がとりあえず計算してみたい! という場合には優しくありません。その点WinmostarはGUIなので操作がしやすいです。計算オプションがデフォルトで設定されているので、とりあえず分子を作って、脳死で「Run」を押せばプログラムが動きます。(本当はちゃんとどのオプションがどういう意味を持っているかは知っておいたほうがいいと思いますが)
あと、複数のソフトウェアを行ったり来たりする必要がないところもポイントだと思います。以前とある研究室でGaussianを動かしたときはまずGaussViewで分子を作成し、そこから分子内の各原子の座標を出力し、それを入力ファイルに貼り付けて計算オプションの設定を記入して……と結構面倒だったのに対し全部が1つのソフトウェアで出来ます。MOPACもGAMESSもGaussianも導入すればWinmostar経由で動かせるので良いですね。
以上、こういう敷居の低さが、初心者がWinmostarを用いる上での利点と言えるのではないでしょうか。なお、単に自分が抱いた印象でしかないので、本格的な機能・性能の比較は詳しい人に聞いたほうが良いと思います。
5.量子化学計算の実践
さて、ここから実際の計算の流れを説明していきます。公式サイトのチュートリアルにその実践例が載っているので、それを参考に分子の配座解析を行ってみます。
エタン分子を考えてみましょう。
詳しい操作手順はチュートリアル(文献[5])に丁寧に書いてあるのと、時間がないので補足情報のみ載せることにしますね。
・Z-Matrixって何?
有機分子の立体的表現はxyz座標表示の他にZ-Matrix表示があります。どのようにして分子を表現しているかというと、
- 最初の原子を原点に置く
- 2番目の原子はx軸方向に置き、1番目の原子との結合距離を記す
- 3番目の原子はxy平面上に置き、1または2番目の原子との結合距離、及び3つの原子からなる結合角を記す
- 4番目以降の原子は既存原子との結合距離、結合角、二面角を記す
Winmostar上では以下の枠内にそのZ-Matrix表示が載っていますね。二面角が0度(重なり形)のエタン分子のZ-matrix表示を詳しく見ていきましょう。
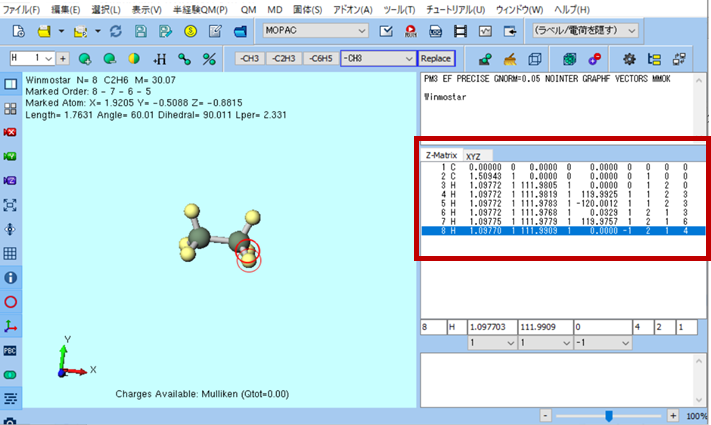

- 「結合距離」はその番号の原子と「結合情報」のNAに記入された番号の原子との距離を表す
- 「結合角」はその番号の原子と「結合情報」のNA、NBに記入された番号の原子となす角を表す
- 「二面角」はその番号の原子と「結合情報」のNA、NBで決まる面と、「結合情報」のNA、NB、NCで決まる面のなす角を表す
- 「結合距離」「結合角」「二面角」のFlagはそれぞれの値を最適化するかを表現しており、1:最適化する、0:固定、-1:連続的に変化する、という意味
・PM3って何?
MOPACの近似計算法の名称です。どのMOPACがどのような計算法に対応しているかはWinmostarのマニュアルから確認することができます。
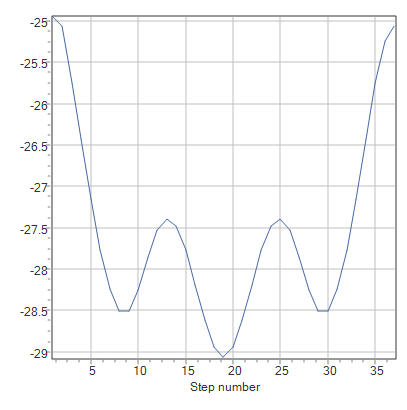
・出力されたグラフがガクガクしている気が……
スキャン計算の設定で、Enter stepのStepを「10」、#Stepsを「36」としたとき、「二面角を10度ずつ36回に渡って変化させる」ことをプログラムに指示していることになります。したがって、グラフ上にプロットされる点は36個になります。なめらかなグラフが欲しい場合は#StepsあたりのStepを小さくすると良いでしょう。
まずはエタン分子の配座解析から。計算をすると以下のようなグラフが出力されました。
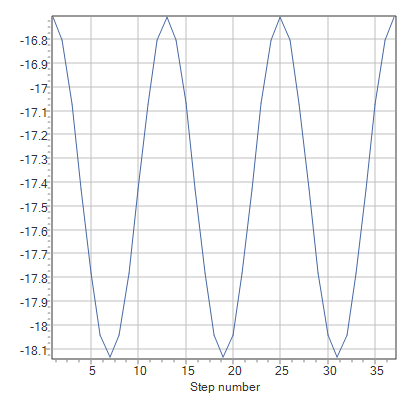
理由は、エタン分子の水素原子同士の反発にあります。二面角が0度のときのエタン分子は重なり形であり、そこから60度ごとに二面角を変えていくとねじれ形と重なり形が交互に変化していきます。このこととグラフを比較してみると重なり形ではエネルギーが極大値を、ねじれ形では極小値をとることがわかります。したがって、ねじれ形のほうが重なり形より安定であり、その原因は水素同士の立体反発にあるのだろうと推測がつくわけです。計算結果からそのエネルギー差は約1.4 kcal/molであることがわかりました。実測値は2.9 kcal/molなので約50%の相対誤差があることになります。意外と大きいですね。
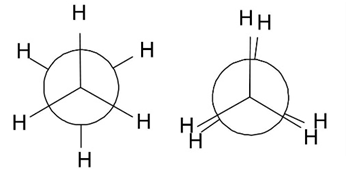
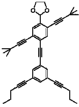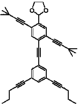
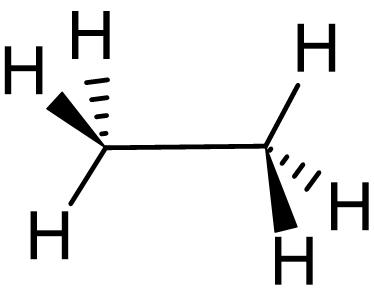
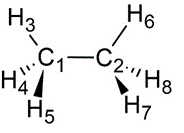
ねじれ形エタン(左)、重なり形エタン(右)のNewman投影式
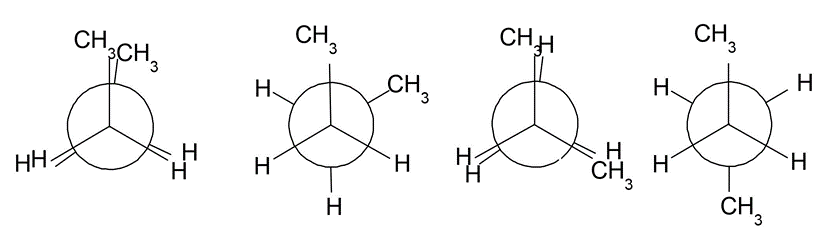
左から、二面角が0度、60度、120度、180度のNewman投影式
他にも様々な分子の二面角を調べることができます。以下に例を挙げるので、ここから先は読者ご自身で計算を実行してみてください。
- イソペンタン、2,3-ジメチルブタン、2-メチルペンタンの配座解析を行ってみましょう。エタン、ブタンの配座解析の結果から、得られるエネルギー図はどのようになるでしょうか? 計算で得られたエネルギー図は予測と合致していましたか?
- 塩素とメチル基の大きさはほぼ同じです。1,2-ジクロロエタンで2つの塩素が重なり形になった配座と、ブタンで2つのメチル基が重なり形になった配座の生成熱を比べてみましょう。生成熱を確認するには、拡張子が.outの出力ファイルをメモ帳で開き、「FINAL HEAT OF FORMATION」を検索してください。(メモ帳の「編集」から検索ができます)このとき最初に出てきた値が二面角が0度のときの生成熱です。検索を続けていくと、1 Step進んだとき、つまり二面角が10度変化したときの値がわかります。どちらの生成熱が高かったですか? 生成熱の差は無視できる程度の差ですか? もし無視できないのであれば、ほぼ同じサイズの置換基なのに生成熱に大きな差が現れたのはなぜなのでしょうか? (ヒント:誘起効果)
6.あとがき
え~、本当は他にもいくつか実践例を用意しておくつもりだったのですが、計画性の無さから駒場祭の直前まで部誌の制作に頭が回らず……中身が物足りない印象を抱いたとしたらごめんなさいです。
検索して調べてみてもWinmostarを使った計算実践例が全然見つからないんですよね。自己流でやろうとしてもいまいちうまく行かなくていくつか没になった計算例が……クロスアビリティ(Winmostarを作った企業)さんが有償で講習を行っているようなので、研究に用いている方々はノウハウをそういうところから得ているのでしょうか。
自分でもあんまし納得がいっていないので、いつかブログにでもまとめるのもありかなあとか思っています。果たしてそんな余裕があるのでしょうか。そもそも教育機関に属している人しか使えないんだから、あんまり人の助けにならないのかもしれませんが。
ではでは。もう一つ記事を書いているのでそちらも是非読んでみてくださいな。
7.参考文献リスト
(最終閲覧日は全て2020/11/19)
[1] 山本悟(1994)「拡張ヒュッケル法のアルミニウム合金への適用」,『軽金属』44(12),p.733-740,軽金属学会.
https://www.jstage.jst.go.jp/article/jilm1951/44/12/44_12_733/_article/-char/ja/
[2] 「MOPACの入手とインストール方法 : PC CHEM BASICS.COM」,
http://pc-chem-basics.blog.jp/archives/1496795.html
[3] 「GAMESS(US)とFireflyの違いって何? : PC CHEM BASICS.COM」,
http://pc-chem-basics.blog.jp/archives/795014.html
[4] 「計算化学:汎関数って何? | Chem-Station (ケムステ)」,
https://www.chem-station.com/blog/2014/12/dft.html
[5] 「Winmostar V10 チュートリアル MOPAC 二面角スキャン」,
https://winmostar.com/jp/tutorials/MOPAC_tutorial_3%28DihedralScan%29.pdf
[6] Maitland Jones,Jr., Steven A. Fleming(2016)『ジョーンズ有機化学 上』(奈良坂紘一ほか訳)東京化学同人.
[7] 染川賢一(2013)『有機分子の分子軌道計算と活用─分子軌道法を用いた有機分子の性質と基本的反応の計算と活用』九州大学出版会.